[
[

Allan-Herndon-Dudley syndrome is a rare congenital disease of thyroid metabolism. It occurs in approximately 1 out of every 70,000 male newborns, and its effects are devastating. Since early 2025, a medication has been approved in Europe that can alleviate symptoms and extend patients’ life expectancy. However, the dosage is complicated.
A team of specialists working with Professor Johannes W. Dietrich from the Department of Diabetology, Endocrinology and Metabolism at the Ruhr University Bochum St. Josef Hospital, Germany—and director of the Center for Rare Endocrine Diseases at the Center for Rare Diseases Ruhr (CeSER)—has published a guideline for treating the disease in the journal Hormone Research in Paediatrics.
Severe symptoms and short life expectancy
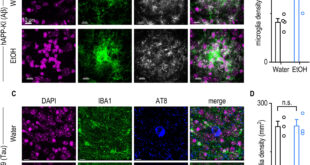
In Allan-Herndon-Dudley syndrome, a mutation in the MCT8 protein severely disrupts the transport of thyroid hormones into and out of certain cell types. The disease is inherited through the X chromosome, is very rare and has devastating consequences for people afflicted by it: severe developmental disorders with consequent major intellectual disability, muscle weakness and cardiac arrhythmias are typical.
“Patients usually never learn how to speak or walk, and, due to an elevated concentration of the thyroid hormone T3, they usually die prematurely from cardiovascular complications,” says Dietrich. “The average life expectancy is 35 years.”
Individual treatment plan
A newly approved medication, the nonclassical thyroid hormone TRIAC, is now available in Europe. This therapy can alleviate some of the symptoms and extend patients’ life expectancy, although the dosage is highly complex.
A working group from Germany and Switzerland developed the new guideline based on extensive research of the literature and its own clinical experience in pediatric neurology and endocrinology. It provides recommendations for a comprehensive, multidisciplinary treatment strategy for patients in all age groups while also considering monitoring of the most important systems and secondary diseases.
“Because the symptoms and long-term courses associated with this disease can vary widely, individual treatment plans for the patients are necessary,” says Dietrich.
More information
Johannes W. Dietrich et al, Clinical and Biochemical Monitoring of Monocarboxylate Transporter 8 Deficiency (Allan-Herndon-Dudley Syndrome) across the Lifespan: Practical Considerations for Multidisciplinary Care, Hormone Research in Paediatrics (2026). DOI: 10.1159/000551857
Key medical concepts


Citation:
New guideline for treating Allan-Herndon-Dudley syndrome (2026, July 8)
retrieved 9 July 2026
from https://medicalxpress.com/news/2026-07-guideline-allan-herndon-dudley-syndrome.html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no
part may be reproduced without the written permission. The content is provided for information purposes only.

Khamrah by Lattafa for Men - 3.4 oz EDP Spray
4% Off
Ghost Sweetheart Eau de Toilette | Pineapple, Jasmine and Sandalwood | Perfume for Women 50 ml
50% Off
Marc Jacobs Dot Eau De Parfum for Women, 100 ml
42% Off
Ted Baker W Eau de Toilette for Her, Fig Leaf, White Peony and Violet Top Notes, Pink Orchid and Raspberry Middle Notes, 75ml
£11.77 (£15.69 / 100 ml) (as of 09/07/2026 03:55 GMT +01:00 - More infoProduct prices and availability are accurate as of the date/time indicated and are subject to change. Any price and availability information displayed on [relevant Amazon Site(s), as applicable] at the time of purchase will apply to the purchase of this product.)
Ted Baker Woman Pink Eau de Toilette Spray Floral Green Feminine Fragrance, Opening Notes are Fresh Peach, Bergamot and Tangerine with Warm Musk, Vanilla and Vetiver Base, 100ml
11% Off
Vera Wang Princess Eau de Toilette - 30 ml

Choco Musk 50ml Eau De Parfum for men and women | Chocolate Musk by Jannat Aromas
17% Off
Christina Aguilera Signature Eau de Parfum (50ml) Floral, Fruity & Exotic Scent, Luxury Fragrance for Women
9% Off
Calvin Klein - Eau De Toilette CKIN2U - Calvin Klein Women, Ladies Perfume, Women's Perfume, Calvin Klein Perfume, Calvin Klein One - 150 ml
5% Off
Jimmy Choo Flash Eau de Parfum, 60 ml (Pack of 1)
3% Off
Fruit of the Loom Men's Heavy T Shirt, White, XL UK
28% Off
ATNKE LED Lighted Beanie Cap,USB Rechargeable Running Hat Ultra Bright 4 LED Waterproof Light Winter Warm Gifts for Men and Women/Pink
17% Off
Men's 1/4 Zip Pullover UK Sale Clearance, Fleece Sweatshirt Casual Jumper Long Sleeve T-shirt Top Stand Collar Sweater Plain Pullover Sports Leisure Workwear Quarter Zip Sweater Lightweight Jumpers
£5.88 (as of 12/11/2025 00:52 GMT +01:00 - More infoProduct prices and availability are accurate as of the date/time indicated and are subject to change. Any price and availability information displayed on [relevant Amazon Site(s), as applicable] at the time of purchase will apply to the purchase of this product.)
Crevice Cleaning Brush, Bathroom Tile Groove Gap Cleaning Brush,Premium Crevice Cleaning Tool Aluminum Support with 15° Angle Magic Brush, Thin Brush for Home Kitchen
19% Off
Wireless Earbuds, Bluetooth 5.3 Headphones in Ear with HiFi Stereo Deep Bass, 4 ENC Noise Cancelling Mic Wireless Earphones 40H Playtime, Bluetooth Earbuds Dual LED Display, IP7 Waterproof, USB-C
42% Off